ГОСТ 11244-76 Нефть. Метод определения потенциального содержания дистиллятных и остаточных масел

ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
НЕФТЬ
МЕТОД ОПРЕДЕЛЕНИЯ
ПОТЕНЦИАЛЬНОГО СОДЕРЖАНИЯ
ДИСТИЛЛЯТНЫХ И ОСТАТОЧНЫХ МАСЕЛ
ГОСТ 11244-76
ГОСУДАРСТВЕННЫЙ КОМИТЕТ
СТАНДАРТОВ
СОВЕТА МИНИСТРОВ СССР
Москва
РАЗРАБОТАН Всесоюзным научно-исследовательским институтом по переработке нефти (ВНИИ НП)
Зам. директора А.Г. Гонсалес
Руководители темы И.Е. Жалнин, З.В. Дриацкая
Исполнители З.В. Масленникова, Н.А. Жмыхова
ВНЕСЕН Министерством нефтеперерабатывающей и нефтехимической промышленности СССР
Член коллегии А.П. Савельев
ПОДГОТОВЛЕН К УТВЕРЖДЕНИЮ Всесоюзным научно-исследовательским институтом стандартизации (ВНИИС)
Директор А.В. Гличев
УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Государственного комитета стандартов Совета Министров СССР от 20 мая 1976 г. № 1238
ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
|
НЕФТЬ Метод определения
потенциального содержания Petroleum. Method for the determination of distillation |
ГОСТ Взамен |
Постановлением Государственного комитета стандартов Совета Министров СССР от 20 мая 1976 г. № 1238 срок введения установлен
с 01.01.1977 г.
до 01.01.1982 г.
Несоблюдение стандарта преследуется по закону
Настоящий стандарт распространяется на нефть и устанавливает метод определения потенциального содержания дистиллятных и остаточных масел.
Сущность метода заключается в разгонке нефти на фракции, депарафинизации, деасфальтенизации и адсорбционном разделении полученных фракций и остатков, последовательном смешении отдельных групп углеводородов и определении в полученных смесях физико-химических показателей.
1. АППАРАТУРА, МАТЕРИАЛЫ И РЕАКТИВЫ
Аппарат АРН-2 по ГОСТ 11011-85.
Воронка Бюхнера № 2 или № 3 по ГОСТ 9147-80, с крышкой, вмонтированная в металлическую баню диаметром 210 мм, высотой 130 мм, обшитую теплоизоляционным материалом.
Чашка выпарительная 7 или 8 по ГОСТ 9147-80.
Колба с тубусом исполнения 1 или 2, вместимостью 500 см3 по ГОСТ 25336-82.
Колбы типа Кн, исполнения 1 или 2, вместимостью 500, 1000 и 2000 см3 из стекла группы ТС или ТХС по ГОСТ 25336-82.
Колбы типа КП, исполнения 1 или 2, вместимостью 250, 500 см3, из стекла группы ТС или ТХС по ГОСТ 25336-82.
Колбы КГУ-2-1-250-29/32 и КГУ-2-1-500-29/32 с насадкой HI -29/32-14/23-14/23, холодильником типа ХПТ-1 или ХПТ-2 длиной 400 или 600 мм, изгибом или алонжем любого типа из стекла ТС или ТХС по ГОСТ 25336-82.
Воронки В-100-150, В-100-200, B -150-230 из стекла группы ТС или ТХС по ГОСТ 25336-82.
Промывалка с резиновой грушей.
Термометры типа ТН-7 или ТН-8 по ГОСТ 400-80.
Термометр ртутный стеклянный лабораторный ТЛ-2 1-Б4 по ГОСТ 215-73.
Колонка адсорбционная (черт. 1) из термически и химически стойкого стекла ТХС, укрепленная на металлической решетке.
К низу колонки присоединяют стеклянный кран, при помощи которого регулируется скорость отбора фильтрата.
Пробирки-приемники из термически и химически стойкого стекла высотой 150 мм и диаметром 36 мм, с меткой на 50 см3 для отбора фракций, вытекающих из колонки. Можно использовать пробирки со шлифом. Растворитель отгоняют непосредственно из этих же пробирок.
Дефлегматор 200-14/23-14/23 ТХС по ГОСТ 25336-82, который присоединяют к пробиркам-приемникам на конусе или при помощи корковой пробки.
Штативы для пробирок длиной 400 мм, шириной 100 мм, в который устанавливают пробирки-приемники. Каждый штатив имеет по 16 отверстий диаметром 38 мм, расположенных в два ряда.
Колонка U -образная (черт. 2) для определения разделяющей способности силикагеля с капельной воронкой ВК-50ХС по ГОСТ 25336-82.
Штативы для укрепления приемников и U -образной колонки.
Приемники для отбора фракций из U -образной колонки представляют собой пробирки высотой (42±1) мм, диаметром (8±0,5) мм, градуированные на 0,2 и 0,3 см3 с погрешностью не более 0,01 см3.
Мановакуумметр U -образный стеклянный по ГОСТ 9933-75.
Рефрактометр типа ИРФ-22 или 454Б.
Шпатель.
Палочки стеклянные с оплавленным концом длиной 150-200 мм.
Насос вакуумный по ГОСТ 14707-82.
Автотрансформатор типа АОСН или другой, позволяющий регулировать обогрев адсорбционной колонки в необходимых пределах.
Баня цилиндрической формы из белой жести внутренним диаметром не менее 250 мм, высотой около 120 мм с тепловой изоляцией для охлаждения продукта и растворителя при депарафинизации.
Электроплитка с закрытой спиралью.
Баня водяная с электронагревом.
Шкаф сушильный, обеспечивающий нагрев (150±5) °С.
Бумага фильтровальная лабораторная по ГОСТ 12026-76.
Метилэтилкетон.
Ацетон по ГОСТ 2603-79.
Бензол нефтяной по ГОСТ 9572-77 или каменноугольный по ГОСТ 8448-78, или бензол по ГОСТ 5955-75.
Толуол нефтяной по ГОСТ 14710-78 или каменноугольный по ГОСТ 9880-76.
Спирт этиловый ректификованный технический по ГОСТ 18300-72.
Изопентан.
a -метилнафталин.
Силикагель марки АСК 0,2-0,5 мм по ГОСТ 3956-76 с разделяющей способностью не менее 40 %, определяемой на контрольной смеси 80 % цетана и 20 % a -метилнафталина (по массе).
Эфир нетролейный марок 40-70 и 70-100.
Двуокись углерода жидкая по ГОСТ 8050-85 или азот газообразный технический по ГОСТ 9293-74 в баллоне, снабженном редукционным вентилем и манометром.
Цетан (гексадекан) эталонный по ГОСТ 12525-85.
Двуокись углерода твердая по ГОСТ 12162-77.
Весы лабораторные с погрешностью взвешивания не более 0,01 г.
(Новая редакция, Изм. № 1).
Адсорбционная колонка
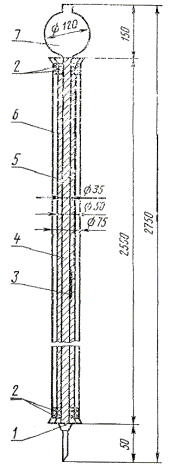
1 - стеклянная вата; 2 - асбест; 3 - термометр; 4 - адсорбционная колонка; 5 - внутренняя трубка муфты со спиралью из нихромовой проволоки; 6 - внешняя трубка муфты; 7 - резервуар для налива испытуемого продукта
Черт. 1
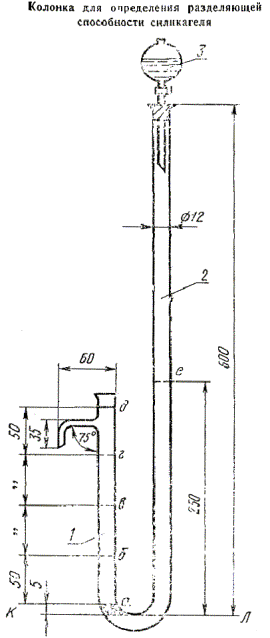
1 - короткое колено U -образной трубки; 2 - длинное колено U -образной трубки; 3 - капельная воронка
Черт. 2
2. ПОДГОТОВКА К АНАЛИЗУ
2.1. Подготовка к депарафинизации
2.1.1. Перед началом депарафинизации приготовляют растворитель смешением 40 объемных долей метилэтилкетона и 60 толуола или 30 объемных долей ацетона и 70 толуола.
2.1.2. Воронку Бюхнера, вмонтированную в металлическую баню, вставляют через пробку в колбу для фильтрования под вакуумом. На дно воронки помещают бумажный фильтр и закрывают ее крышкой. В баню наливают этиловый спирт, устанавливают термометр типа ТН-8 и охлаждают воронку.
2.1.3. Колбу для фильтрования соединяют вакуумной резиновой трубкой через промежуточную колбу с вакуумным насосом. К вакуумной трубке через тройник присоединяют ртутный вакуумметр, а через другой тройник - кран, соединенный с атмосферой.
2.1.4. Для охлаждения растворенной навески испытуемого продукта и растворителя готовят вторую баню, которую наполняют этиловым спиртом и устанавливают термометр типа ТН-8.
2.2. Подготовка к адсорбционному разделению
2.2.1. Силикагель помещают в фарфоровую чашу и выдерживают в сушильном шкафу 12 ч три 150±5 °С, периодически помешивая.
Высушенный силикагель в горячем состоянии переносят в нагретую стеклянную колбу и плотно закрывают резиновой пробкой, взвешивают.
(Измененная редакция, Изм. № 1 ).
2.2.2. Определение разделяющей способности силикагеля, подготовленного по п. 2.2.1 .
В нижнюю часть короткого колена и отвод колена U -образной колонки (черт. 2) помещают стеклянную вату для удержания силикагеля. Силикагель массой 12-15 г насыпают небольшими порциями в короткое колено и уплотняют до метки «д» непрерывным постукиванием по колонке деревянной палочкой. Массу силикагеля определяют по разности массы колбы с силикагелем до и после наполнения трубки.
Контрольную смесь из капельной воронки вносят по каплям в длинное колено трубки со скоростью, обеспечивающей пропитку силикагеля контрольной смесью не менее чем за 1 ч, при этом смесь должна последовательно пройти через каждые 15 мин отметки а, б, в, г, д на коротком колене.
Фракции отбирают через боковой отвод в приемники по 0,3 см3 в каждый со скоростью 0,3 см3/мин.
Скорость регулируют высотой столба жидкости в высоком колене.
Отбирают 20-25 фракций, количество которых обеспечивает появление a -метилнафталина.
С помощью рефрактометра
определяют показатель преломления ( ![]() ) в каждой фракции. Повышение показателя преломления на
0,0005 указывает на появление a -метилнафталина.
) в каждой фракции. Повышение показателя преломления на
0,0005 указывает на появление a -метилнафталина.
Разделяющую способность силикагеля (А) в процентах вычисляют по формуле
![]() ,
,
где 0,3 - объем продукта в каждом приемнике, см3;
0,774 - плотность цетана, г/см3;
п - количество фракций, свободных от a -метилнафталина;
т - масса силикагеля, г.
(Измененная редакция, Изм. № 1 ).
2.2.3. Адсорбционную колонку (черт. 1 ) заполняют силикагелем, подготовленным по п. 2.2.1 . Силикагель насыпают небольшими порциями и уплотняют непрерывным постукиванием по колонке деревянной палочкой. Количество силикагеля, помещенного в адсорбционную колонку, определяют по разности массы колбы с силикагелем до и после наполнения колонки.
(Измененная редакция, Изм. № 1 ).
2.2.4. Силикагель регенерируют после каждого адсорбционного разделения для многократного его использования. Для этого к верхней части резервуара присоединяют каучуковую трубку для подачи инертного газа из баллона через редукционный вентиль. Одновременно включают обогрев колонки и температуру медленно повышают до тех пор, пока не прекратится выделение паров растворителя из колонки.
Пары растворителя отводят под тягу через ловушку, присоединенную внизу колонки.
Затем температуру в колонке повышают до 150 °С и поддерживают ее в течение 2-3 ч в токе инертного газа. После этого обогрев колонки выключают и силикагель охлаждают также в токе инертного газа. Когда температура в колонке снизится до температуры окружающей среды, подачу инертного газа прекращают.
После каждых 10 опытов проверяют разделяющую способность силикагеля в соответствии с п. 2.2.2.
При снижении активности на 3-4 единицы силикагель заменяют.
Регенерацию силикагеля можно проводить вне колонки, промывая его горячей дистиллированной водой и высушивая в сушильном шкафу, как указано в п. 2.2.1.
2.3. (Исключен, Изм. № 1 ).
3. ПРОВЕДЕНИЕ АНАЛИЗА
3.1. Отбор фракций нефти
3.1.1. Испытуемую нефть перегоняют в аппарате АРН-2 по ГОСТ 11011-85 и отбирают для испытаний фракции, выкипающие в пределах 300 - 400 °С, 400 - 450 °С, 450 - 500 °С, и остаток, или 300 - 350 °С, 350 - 400 °С, 400 - 450 °С, 450 - 500 °С и остаток, или 300 - 350 °С, 350 - 420 °С, 420 - 500 °С и остаток, или 300 - 350 °С, 350 - 450 °С, 450 - 500 °С и остаток, или остаток выше 350 °С.
(Новая редакция, Изм. № 1 ).
3.1.2. Порядок испытаний дистиллятных фракций и остатков представлен на черт. 3- 5.
(Новая редакция, Изм. № 1 ).
3.2. Деасфальтенизация
3.2.1. Деасфальтенизации подвергают остатки, полученные по п. 3.1.1. (черт. 4, 5).
(Измененная редакция, Изм. № 1 ).
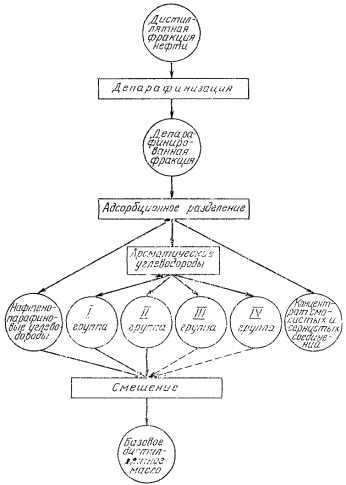
Черт. 3
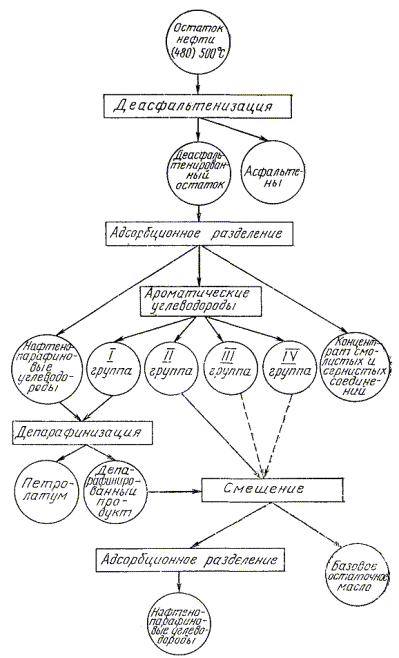
Черт. 4
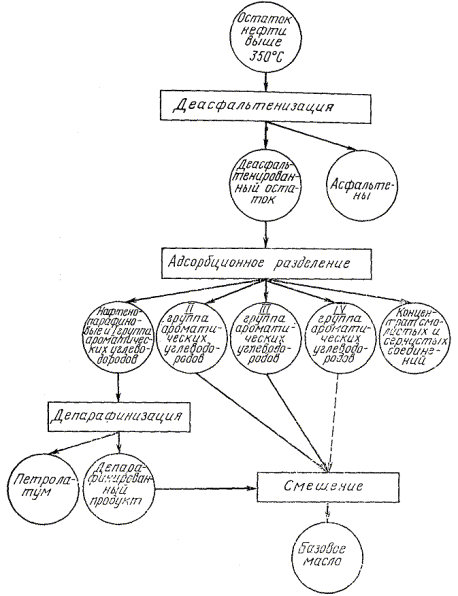
Черт. 5
Если содержащие асфальтенов в нефти не более 2,5 %, то адсорбционное разделение остатка выше 350 °С проводят без предварительной деасфальтенизации.
3.2.2. В коническую колбу вместимостью 2000 см3 помещают 100 г остатка после перегонки нефти и постепенно приливают при тщательном перемешивании 10-кратное количество изопентана или петролейного эфира (марка 40-70 °С), после чего оставляют раствор не менее чем на 3 ч для выделения асфальтенов.
(Измененная редакция, Изм. № 1 ).
3.2.3. Выпавшие асфальтены отфильтровывают через бумажный фильтр и промывают петролейным эфиром (марка 70-100 °С) с целью более полного извлечения масла.
3.2.4. Фильтрат сливают в предварительно взвешенную колбу типа КП. В горло колбы вставляют на пробке капиллярную трубку, через которую подводят углекислый газ. Ставят колбу в водяную баню с электронагревом, соединяют отводную трубку колбы с холодильником и отгоняют растворитель в токе углекислого газа.
Затем колбу с деасфальтенированным остатком охлаждают до температуры окружающей среды и взвешивают.
3.2.5. Выход деасфальтенированного остатка (Х1) в процентах вычисляют по формуле
![]() ,
,
где m 1 - масса остатка, взятого для деасфальтенизации, г;
m 2 - масса деасфальтенированного остатка, г.
3.3. Депарафинизация
3.3.1. Депарафинизации подвергают дистиллятные фракции, полученные по п. 3.1.1, смесь нафтено-парафиновых (циклано-алкановых) и I группы ароматических (ареновых) углеводородов, полученных после адсорбционного разделения остатков выше 350 °С и (480) 500 °С (см. черт. 3, 4, 5).
Если содержание парафина в нефти не более 1,5 %, адсорбционное разделение дистиллятных фракций проводят без депарафинизации.
(Измененная редакция, Изм. № 1 ).
3.3.2. В коническую колбу вместимостью 500 см3 помещают 100 г анализируемого нефтепродукта.
Дистиллятные фракции нагревают в колбе на водяной бане до 50-60 °С (до полного расплавления парафина), затем охлаждают до температуры окружающей среды.
Смесь нафтено-парафиновых и I группы ароматических углеводородов нагревают в колбе до 50-60 °С, добавляют половину требуемого растворителя (п. 1.1), количество которого определяется по табл. 1, после чего раствор вновь нагревают до полного растворения парафинов, затем охлаждают при перемешивании до температуры окружающей среды.
(Измененная редакция, Изм. № 1 ).
3.3.3. Нефтепродукт, подготовленный по п. 3.3.2 , помещают в баню и добавлением твердой углекислоты при перемешивании снижают температуру бани со скоростью 1-2 °С/мин.
Таблица 1
|
Наименование нефтепродукта |
Соотношение нефтепродукта и растворителя |
|
Дистиллятная фракция, полученная из нефти с содержанием парафина, %: |
|
|
до 6,5 |
1:3 |
|
св. 6,5 |
1:4 |
|
Смесь нафтено-парафиновых с I группой ароматических углеводородов, полученная при адсорбционном разделении остатков из нефтей с содержанием парафина, %: |
|
|
до 6,5 |
1:4 |
|
св. 6,5 |
1:6 |
Одновременно в баню помещают колбы с растворителем в количестве, необходимом для депарафинизации и промывки фильтра. К дистиллятной фракции после выпадения кристаллов парафина добавляют половину требуемого количества растворителя, взятого в соответствии с табл. 1 и охлажденного до температуры продукта, и охлаждают при перемешивании.
При температуре раствора минус 27-28 °С, колбы с раствором выдерживают 10-12 мин, затем добавляют вторую часть растворителя, охлажденного до минус 27-28 °С. За это время температуру охладительной смеси в бане, в которую вмонтирована воронка Бюхнера, снижают до минус 32-33 °С.
(Измененная редакция, Изм. № 1 ).
3.3.4. Включают вакуумный насос, закрывают кран, соединяющий колбу для фильтрования с атмосферой, создают вакуум 0,4 МПа (300 мм рт. ст.), смачивают фильтр в воронке охлажденным растворителем, подготовленным для промывки осадка на фильтре, и выливают на фильтр испытуемый раствор с выпавшим парафином. Стенки колбы смывают частью охлажденного растворителя, который сливают на фильтр.
Осадок петролатума или гача на фильтре промывают оставшимся охлажденным растворителем, разравнивая лепешку шпателем для равномерного распределения растворителя по поверхности лепешки и во избежание образования трещин.
Соотношение нефтепродукта, взятого для депарафинизации, и растворителя для промывки фильтра 1:1.
По окончании промывки и фильтрования открывают кран, соединяющий систему с атмосферой, и выключают вакуумный насос.
(Измененная редакция, Изм. № 1 ).
3.3.5. Для удаления следов масла из осадка его подвергают дополнительной обработке. Для этого осадок снимают с фильтра шпателем, разбавляют его охлажденным до минус 28 °С растворителем (соотношение продукта и растворителя 1:1-1:1,5 в пересчете на исходный нефтепродукт), снова отфильтровывают при температуре, указанной в п. 3.3.3, и промывают растворителем в количестве 1:1.
По окончании промывки и фильтрования открывают кран, соединяющий систему с атмосферой, выключают вакуумный насос.
(Измененная редакция, Изм. № 1 ).
3.3.6. Шпателем снимают с фильтра петролатум, выпаривают растворитель, сушат в сушильном шкафу при (100±5) °С до постоянной массы, затем взвешивают и определяют температуру плавления.
(Измененная редакция, Изм. № 1 ).
3.3.7. Фильтрат из колбы для фильтрования под вакуумом сливают в предварительно взвешенную колбу типа КГУ, соединенную с насадкой, холодильником и изгибом, в горло колбы вставляют на пробке капиллярную трубку, подводящую азот или углекислый газ, в тубус вставляют термометр типа ТН-7, ставят колбу в колбонагреватель и соединяют отводную трубку колбы с холодильником.
Растворитель отгоняют в токе инертного газа до температуры, не превышающей 140 °С в жидкости.
После отгона растворителя колбу с депарафинированным маслом охлаждают до температуры окружающей среды и взвешивают.
(Измененная редакция, Изм. № 1 ).
3.3.8. Массовую долю петролатума (Х2) в процентах вычисляют по формуле
![]() ,
,
где m 3 - масса петролатума после отгона растворителя, г;
m 4 - масса продукта, подвергнутого депарафинизации, г.
Массовую долю депарафинированного масла (Х3) в процентах вычисляют по формуле
![]() ,
,
где m 4 - масса продукта, подвергнутого депарафинизации, г;
m 5 - масса масла после депарафинизации и отгона растворителя, г.
(Новая редакция, Изм. № 1 ).
3.4. Адсорбционное разделение
3.4.1. Адсорбционному разделению подвергают депарафинированные фракции (п. 3.3), деасфальтенированные остатки (п. 3.2) и депарафинированную смесь нафтено-парафиновых и ароматических углеводородов, полученную после адсорбционного разделения остатка (см. черт. 4). Соотношение продукта и силикагеля 1:10, для высокосмолистых - 1:15.
3.4.2. Адсорбционную колонку, подготовленную как указано в п. 2.2.3, пропитывают петролейным эфиром (марка 70-100 °С), для чего в резервуар помещают 1000-1500 см3 петролейного эфира.
(Измененная редакция, Изм. № 1 ).
3.4.3. 50-100 г нефтепродукта растворяют в петролейном эфире (марка 70-100 °С). Соотношение массы нефтепродукта и петролейного эфира 1:3.
3.4.4. Как только петролейный эфир полностью смочит силикагель и сверху колонки останется столб петролейного эфира высотой 2-5 мм, в колонку помещают раствор нефтепродукта, подготовленный по п. 3.4.3. При разделении деасфальтенированных остатков перед помещением раствора нефтепродукта нагревают колонку до 35-45 °С.
3.4.5. С помощью крана внизу колонки устанавливают скорость отбора раствора 3,0-3,5 см3/мин.
3.4.6. Под колонку подставляют колбу вместимостью 1000 см3 для отбора чистого петролейного эфира.
3.4.7. После того, как раствор анализируемого нефтепродукта почти полностью войдет в силикагель, не допуская высыхания верхнего слоя силикагеля, в резервуар колонки порциями помещают 2500 см3 петролейного эфира. Каждую последующую порцию добавляют, не давая высыхать верхнему слою силикагеля.
Указанные в этом пункте и п. 3.4.8 количества растворителя даны в расчете на загрузку 50-100 г исследуемого нефтепродукта.
(Измененная редакция, Изм. № 1 ).
3.4.8. После того, как весь петролейный эфир войдет в силикагель, для более четкого разделения отдельных групп ароматических углеводородов и смолистых веществ, в колонку помещают последовательно смеси растворителей, указанных в табл. 2 .
Таблица 2
|
Состав смеси, % |
Количество смеси , см3 |
|
|
Бензол |
Петролейный эфир |
|
|
5 |
95 |
1500 |
|
15 |
85 |
1500 |
|
100 |
0 |
500 |
При адсорбционном разделении деасфальтенированного остатка объем смеси (15 % бензола и 85 % петролейного эфира) увеличивают до 2000 см3.
(Измененная редакция, Изм. № 1 ).
3.4.9. Для полной десорбции смолистых веществ после того, как последняя часть смеси бензола и петролейного эфира войдет в силикагель, в резервуар колонки помещают 500 см3 спиртобензольной смеси в соотношении 1:1.
Полное вытеснение спирто-бензольной смеси осуществляется 400-800 см3 петролейного эфира до выхода из адсорбционной колонки неокрашенных соединений.
(Измененная редакция, Изм. № 1 ).
3.4.10. При заполнении колонки петролейным эфиром, раствором исходного продукта и последующей десорбции из адсорбционной колонки сначала вытесняется чистый петролейный эфир, затем последовательно растворы отдельных групп углеводородов и концентрат смолистых и сернистых соединений.
Объем вытесненного чистого петролейного эфира обычно составляет около 70 % растворителя, израсходованного на заполнение колонки, и он может быть повторно использован без перегонки.
(Измененная редакция, Изм. № 1 ).
3.4.11. Растворы, вытесненные снизу колонки, после отбора около 700 см3 чистого петролейного эфира, собирают в отдельные пробирки-приемники по 50 см3. От каждой полученной фракции отгоняют растворитель.
Для этого в приемник с помощью корковой пробки вставляют дефлегматор и трубку для пропускания инертного газа. Приемник помещают в водяную баню и соединяют дефлегматор с холодильником. Отгоняют растворитель при температуре бани 90-100 ° C в токе инертного газа. При перегонке сильно разбавленных растворов во избежание потери выделенных при адсорбции фракций смежные фракции последовательно объединяют и отгоняют в одном и том же приемнике до накопления 2-3 г фракции.
(Измененная редакция, Изм. № 1 ).
3.4.12. Фракции, полученные после отгона растворителя, взвешивают, вычисляют их массовую долю в процентах и определяют показатель преломления и дисперсию.
(Новая редакция, Изм. № 1 ).
3.5. Определение содержания групп углеводородов и смолистых веществ, полученных при адсорбционном разделении
Полученные при адсорбционном разделении фракции смешивают с целью выделения групп, указанных ниже.
3.5.1. К нафтено-парафиновым углеводородам относят фракции с показателем преломления не более 1,49 и величиной дисперсии не выше 85.
3.5.2. Ароматические углеводороды (смесь ароматических углеводородов и сернистых соединений) разбивают на четыре группы по условно принятым пределам значений показателя преломления.
(Измененная редакция, Изм. № 1 ).
3.5.2.1. К I группе ароматических углеводородов относят фракции, полученные после отбора нафтено-парафиновых углеводородов, с показателем преломления свыше 1,49 до 1,53.
3.5.2.2. Ко II группе ароматических углеводородов относят фракции с показателем преломления свыше 1,53 до 1,55.
3.5.2.3. К III группе ароматических углеводородов относят фракции с показателем преломления свыше 1,55 до 1,59.
3.5.2.4. К IV группе ароматических углеводородов относят фракции с показателем преломления свыше 1,59.
Для отдельных нефтей после отбора фракций с показателем преломления свыше 1,59 наблюдается понижение значения показателя преломления за счет увеличения содержания сернистых соединений. Такие фракции относят к IV группе ароматических углеводородов.
3.5.3. К группе смолистых и сернистых соединений относят фракции, у которых из-за темного цвета не представляется возможным определить показатель преломления.
3.6. Составление смесей из фракций адсорбционного разделения с целью установления потенциального содержания базовых дистиллятных масел
3.6.1. В нафтено-парафиновых углеводородах определяют плотность по ГОСТ 3900-85 , показатель преломления, удельную дисперсию, вязкость при 40 ° С и 100 ° С по ГОСТ 33-82 , индекс вязкости по ГОСТ 25371-82 и температуру застывания по ГОСТ 20287-74.
(Новая редакция, Изм. № 1 ).
3.6.2. Оставшиеся после анализа нафтено-парафиновые углеводороды взвешивают и к ним добавляют I группу ароматических углеводородов.
Количество I группы ароматических углеводородов ( aI ) в граммах, которое необходимо добавить к нафтено-парафиновым углеводородам, вычисляют по формуле
![]() ,
,
где н - количество нафтено-парафиновых углеводородов, оставшееся после анализа, г;
Н - содержание нафтено-парафиновых углеводородов, полученных при адсорбционном разделении исследуемого продукта, %;
А I - содержание I группы ароматических углеводородов, полученных при адсорбционном разделении исследуемого, продукта, %.
В полученной смеси определяют показатели, указанные в п. 3.6.1, и дополнительно коксуемость по ГОСТ 19932-74.
3.6.3. Оставшуюся после анализа смесь нафтено-парафиновых углеводородов и I группы ароматических углеводородов (п. 3.6.1 ) взвешивают и к ней добавляют II группу ароматических углеводородов, полученных по п. 3.5.2.2 .
Количество II группы ароматических углеводородов (а II ) в граммах, которое надо добавить к смеси (н+а I ), вычисляют по формуле
![]() ,
,
где н+а I - количество смеси нафтено-парафиновых углеводородов с I группой ароматических углеводородов, оставшееся после анализа, г;
Н+А I - содержание смеси нафтено-парафиновых углеводородов с I группой ароматических углеводородов, полученных при адсорбционном разделении исследуемого продукта, %;
AII - содержание II группы ароматических углеводородов, полученных при адсорбционном разделении исследуемого продукта, %.
Полученную смесь анализируют по показателям п. 3.6.1.
(Измененная редакция, Изм. № 1 ).
3.6.4. Оставшуюся после анализа смесь взвешивают и к ней прибавляют III группу ароматических углеводородов, полученных по п. 3.5.2.3 .
Количество III группы ароматических углеводородов (а III ) в граммах, которое надо добавить к смеси (н+а I +а II ), вычисляют по формуле
![]() ,
,
где н+а I +а II - количество смеси, оставшееся после анализа по п. 3.6.3, г;
Н+А I +А II - содержание смеси нафтено-парафиновых углеводородов с I и II группами ароматических углеводородов, полученных при адсорбционном разделении исследуемого продукта, %;
А III - содержание III группы ароматических углеводородов, полученных при адсорбционном разделении исследуемого продукта, %.
Полученную смесь анализируют по показателям п. 3.6.1.
(Измененная редакция, Изм. № 1 ).
3.6.5. Оставшуюся после анализа смесь взвешивают и к ней прибавляют IV группу ароматических углеводородов, полученных по п. 3.5.2.4 .
Количество IV группы ароматических углеводородов ( aIV ) в граммах, которое надо добавить к смеси (н+ aI + aII +а III ) , вычисляют по формуле
![]() ,
,
где н+ aI +а II + aIII - количество смеси, оставшееся после анализа по п. 3.6.4, г;
H + AI +А II + AIII - содержание смеси нафтено-парафиновых с I , II и III группами ароматических углеводородов, полученных при адсорбционном разделении исследуемого продукта, %;
AIV - содержание IV группы ароматических углеводородов, полученных при адсорбционном разделении исследуемого продукта, %.
Полученную смесь анализируют по показателям п. 3.6.1.
(Измененная редакция, Изм. № 1 ).
3.7. Составление смесей из фракций адсорбционного разделения с целью установления по тенциального содержания базовых остаточных масел
3.7.1. Смесь нафтено-парафиновых углеводородов и I группы ароматических углеводородов после депарафинизации, полученных по п. 3.3, (см. черт. 4, 5), анализируют в соответствии с п. 3.6.1.
(Измененная редакция, Изм. № 1 ).
3.7.2. К оставшейся смеси прибавляют последовательно II , III и IV группы ароматических углеводородов (см. черт. 4 , 5 ), как указано в пп. 3.6.3 , 3.6.4 и 3.6.5 , и определяют показатели, указанные в п. 3.6.1 .
(Измененная редакция, Изм. № 1 ).
3.7.3. Оставшуюся смесь по п. 3.7.2 подвергают повторному адсорбционному разделению по п. 3.4 с целью выделения и анализа группы нафтено-парафиновых углеводородов по п. 3.6.1 (см. черт. 4).
4. ОБРАБОТКА РЕЗУЛЬТАТОВ
4.1. Результаты анализа записывают в таблицу и выражают графически в виде зависимости свойств масел от глубины адсорбционного разделения исследуемого продукта.
Пример записи результатов анализа дан в справочном приложении (см. табл. 2 и чертеж).
4.2. Общее потенциальное содержание дистиллятных и остаточных базовых масел в нефти рассчитывают по содержанию масел с одинаковым индексом вязкости (ИВ), определенному по кривым зависимости ИВ масел от глубины адсорбционного разделения фракций и остатка. При этом следует учитывать, что масла не должны состоять только из нафтено-парафиновых углеводородов и не должны полностью содержать IV группу ароматических углеводородов.
Пример. Из дистиллята, выкипающего в пределах 300-400 ° С, получено, считая на нефть, базового масла с ИВ 85 - 14 %, из дистиллята 400-450 ° С - 5,6 %, из дистиллята 450-500 ° С - 3,7 %, из остатка выше 500 ° С - 2,7 %. Общее потенциальное содержание базовых дистиллятных и остаточных масел с ИВ 85 составляет 14+5,6+3,7+2,7=26 %, считая на нефть.
За результат исследования принимают среднее арифметическое результатов двух параллельных определений.
(Новая редакция, Изм. № 1 ).
4.3. Допускаемые расхождения между параллельными определениями не должны превышать величин, указанных в табл. 3.
Таблица 3
|
Выход базовых масел (в пересчете на нефть), % |
Допускаемое расхождение, % |
|
До 2 |
0,2 |
|
Св. 2 до 5 |
0,3 |
|
Св. 5 |
0,5 |
ПРИЛОЖЕНИЕ
Справочное
ПРИМЕР ЗАПИСИ РЕЗУЛЬТАТОВ АДСОРБЦИОННОГО РАЗДЕЛЕНИЯ ДЕПАРАФИНИРОВАННОГО ДИСТИЛЛЯТА И ГРУППИРОВКИ ПОЛУЧЕННЫХ ФРАКЦИЙ
При адсорбционном разделении депарафинированного дистиллята, выкипающего в пределах 450-500 °С, и анализе отдельных фракций получены данные, на чертеже и указанные в табл. 1, 2.
Кривые зависимости свойств масел от глубины адсорбционного разделения ди стиллята 450-500 °С
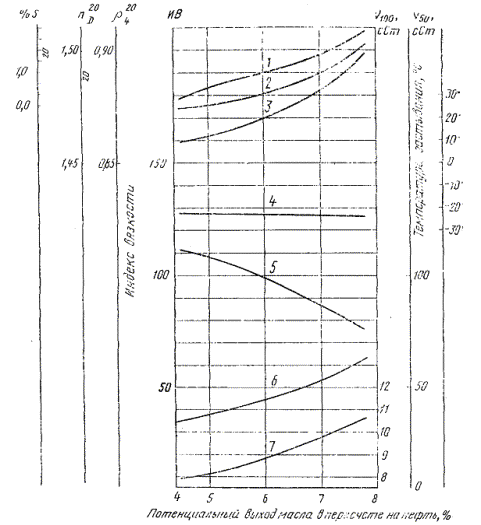
1 - содержание серы, %; 2 - показатель
преломления ![]() ; 3 - плотность при 20 ° С; 4 - температура застывания, ° С; 5 - индекс вязкости; 6 - кинематическая вязкость при
50 ° С; 7 - кинематическая
вязкость при 100 °С
; 3 - плотность при 20 ° С; 4 - температура застывания, ° С; 5 - индекс вязкости; 6 - кинематическая вязкость при
50 ° С; 7 - кинематическая
вязкость при 100 °С
Таблица 1
|
Содержание фракции, % |
Показатель преломления |
Дисперсия |
Группа углеводородов |
Содержание групп углеводородов на дистиллят, % |
||
|
отдельной |
суммарной |
депарафинированный |
исходный |
|||
|
2,7 |
2,7 |
1,4727 |
- |
Нафтено-парафиновые углеводороды |
58,5 |
52,4 |
|
20,3 |
23,0 |
1,4787 |
- |
|||
|
16,8 |
39,8 |
1,4794 |
- |
|||
|
9,5 |
49,3 |
- |
- |
|||
|
5,7 |
55,0 |
1,4805 |
84 |
|||
|
3,1 |
58,1 |
1,4815 |
84 |
|||
|
0,4 |
58,5 |
1,4840 |
85 |
|||
|
0,3 |
58,8 |
1,4913 |
98 |
I группа ароматических углеводородов |
15,1 |
13,5 |
|
2,0 |
60,8 |
1,4988 |
- |
|||
|
3,8 |
64,6 |
1,5017 |
- |
|||
|
5,1 |
69,7 |
1,5128 |
- |
|||
|
1,9 |
71,6 |
1,5140 |
- |
|||
|
1,4 |
73,0 |
1,5175 |
- |
|||
|
0,6 |
73,6 |
1,5260 |
- |
|||
|
0,6 |
74,2 |
1,5302 |
- |
II группа ароматических углеводородов |
6,7 |
6,0 |
|
3,4 |
77,6 |
1,5386 |
- |
|||
|
1,7 |
79,3 |
1,5390 |
- |
|||
|
0,6 |
79,9 |
1,5421 |
- |
|||
|
0,4 |
80,3 |
1,5473 |
- |
|||
|
0,2 |
80,5 |
1,5540 |
- |
III группа ароматических углеводородов |
5,7 |
5,1 |
|
0,8 |
81,3 |
1,5560 |
- |
|||
|
1,6 |
82,9 |
1,5584 |
- |
|||
|
1,9 |
84,8 |
1,5609 |
- |
|||
|
1,1 |
85,9 |
1,5773 |
- |
|||
|
0,1 |
86,0 |
1,5822 |
- |
|||
|
0,8 |
86,8 |
1,5925 |
- |
IV группа ароматических углеводородов |
11,8 |
10,6 |
|
4,6 |
91,4 |
1,5936 |
- |
|||
|
4,2 |
95,6 |
1,5974 |
- |
|||
|
1,2 |
96,8 |
1,5980 |
- |
|||
|
1,0 |
97,8 |
1,5990 |
- |
|||
|
2,2 |
100,0 |
Определить нельзя (темный цвет) |
- |
Концентрат смолистых и сернистых соединений |
2,2 |
2,0 |
Таблица 2
|
Наименование группы |
Содержание масла, % |
Плотность относительная |
Показатель преломления |
Удельная дисперсия SFC |
Кинематическая вязкость, сСт (м2/с) |
Индекс вязкости |
Температура застывания, °С |
Сера, % |
|
||
|
50 °С |
100 °С |
||||||||||
|
на дистиллят |
на нефть |
|
|||||||||
|
Исходный дистиллят 450-500 °С |
100,0 |
8,7 |
0,8903 |
1,4989 |
- |
52,94 (52,94∙10-6) |
9,50 (9,50∙10-6) |
- |
41 |
1,17 |
|
|
То же, после депарафинизации* |
89,6 |
7,8 |
0,8982 |
1,5020 |
- |
63,00 (63,00∙10-6) |
10,50 (10,50∙10-6) |
76 |
минус 24 |
1,32 |
|
|
Нафтено-парафиновые углеводороды |
52,4 |
4,5 |
0,8587 |
1,4735 |
100 |
34,71 (34,71∙10-6) |
8,02 (8,02∙10-6) |
111 |
минус 22 |
0,16 |
|
|
То же + I группа ароматических углеводородов |
65,9 |
5,7 |
0,8670 |
1,4785 |
111 |
41,54 (41,54∙10-6) |
8,59 (8,59∙10-6) |
102 |
минус 23 |
0,48 |
|
|
То же + II группа ароматических углеводородов |
71,9 |
6,2 |
0,8718 |
1,4823 |
120 |
45,55 (45,55∙10-6) |
8,94 (8,94∙10-6) |
96 |
минус 23 |
0,63 |
|
|
То же + III группа ароматических углеводородов |
77,0 |
6,7 |
0,8773 |
1,4855 |
134 |
50,40 (50,40∙10-6) |
9,48 (9,48∙10-6) |
90 |
минус 23 |
0,80 |
|
|
То же + IV группа ароматических углеводородов |
87,6 |
7,6 |
0,8925 |
1,4968 |
200 |
90,50 (60,50∙10-6) |
10,32 (10,32∙10-6) |
79 |
минус 24 |
1,10 |
|
* Содержание гача 10,4 %, считая на дистиллят, или 0,9 %, считая на нефть, температура плавления гача 59 °С.
Пользуясь графиком, находим, например, что содержание базового дистиллятного масла с кинематической вязкостью при 50 °С 47,00 сСт (47 ∙ 10-6 м2/с) и индексом вязкости 95 составляет 6,3 %, считая на нефть, а с вязкостью 50,40 сСт (50,40 ∙ 10-6 м2/с) и индексом вязкости 90 составляет 6,7 %, считая на нефть.
СОДЕРЖАНИЕ
|
1. Аппаратура, материалы и реактивы .. 2 2. Подготовка к анализу . 4 3. Проведение анализа . 6 4. Обработка результатов . 14 Приложение Пример записи результатов адсорбционного разделения депарафинированного дистиллята и группировки полученных фракций . 14 |